Congenital Adrenocortical Tumor in an Asymptomatic Neonate with Positive Newborn Screen for Congenital Adrenal Hyperplasia: A Case Report
Keywords:
Adrenocortical tumor, newborn screening, 17-hydroxyprogesterone, congenital adrenal hyperplasia, Li-Fraumeni syndrome
Abstract
Adrenocortical tumors (ACTs) are rare in childhood, particularly in the neonatal period. ACTs in children usually secrete hormones that can cause virilization, precocious puberty, or Cushing syndrome. However, in many cases the diagnosis is delayed because of the generally healthy appearance of the child and the lack of a palpable abdominal mass. ACTs can also be misdiagnosed as congenital adrenal hyperplasia (CAH) especially if 17-hydroxyprogesterone (17-OHP) is elevated. We describe a case of neonatal ACT diagnosed in an asymptomatic newborn with positive/elevated 17-OHP on newborn screening. On examination there were no signs of virilization, Cushing syndrome, hemihypertrophy, or hypertension, and there was no palpable abdominal mass. While awaiting confirmatory results of an elevated 17-OHP level, an abnormally low adrenocorticotropic hormone level was noted. Abdominal ultrasound, obtained to evaluate the adrenal glands, showed a right adrenal mass with peripheral calcification. MRI revealed a 3.6 x 3.5 x 3.4 cm heterogeneously enhancing ovoid mass in the right adrenal gland. Diagnosis of an ACT was also confirmed by laboratory data, and histopathology of the tumor. Surgical resection was safely performed with perioperative steroid replacement. Genetic analysis of CYP21A2 and CYP11B1 genes was negative. A germ-line point mutation in the TP53 region was discovered, suggesting Li-Fraumeni syndrome. ACTs should be considered in the differential diagnosis of neonates with elevated 17-OHP detected by newborn screening for CAH. Adrenal ultrasound is recommended in cases of significantly abnormal CAH screen results to rule out or treat adrenal neoplasms.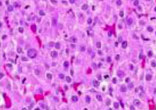
Published
2017-02-14
How to Cite
Thao Hoang, P., Eric, T., Mace, J., & Hathout, E. (2017). Congenital Adrenocortical Tumor in an Asymptomatic Neonate with Positive Newborn Screen for Congenital Adrenal Hyperplasia: A Case Report. International Journal of Integrative Pediatrics and Environmental Medicine, 3, 1-4. https://doi.org/10.36013/ijipem.v3i0.30
Issue
Section
Articles
Policy for Articles with Open Access
Authors who publish with this journal agree to the following terms:
Authors retain copyright and grant the journal right of first publication with the work simultaneously licensed under a Creative Commons Attribution License that allows others to share the work with an acknowledgement of the work's authorship and initial publication in this journal.
Authors are permitted and encouraged to post links to their work online (e.g., in institutional repositories or on their website) prior to and during the submission process, as it can lead to productive exchanges, as well as earlier and greater citation of published work.




3.jpg)
